Pharmacologic Category: Calcium Channel Blocker; Lipid Regulating Agent.
Pharmacology: Pharmacodynamics: Mechanism of Action: Amlodipine besilate/Atorvastatin calcium (Norvasc Protect) combines two mechanisms of action: the dihydropyridine calcium antagonist (calcium ion antagonist or slow-channel blocker) action of amlodipine and the HMG-CoA reductase inhibition of atorvastatin. The amlodipine component of amlodipine/atorvastatin inhibits the transmembrane influx of calcium ions into vascular smooth muscle and cardiac muscle. The atorvastatin component of amlodipine/atorvastatin is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts HMG-CoA to mevalonate, a precursor of sterols, including cholesterol.
Clinical Studies of Combined Amlodipine and Atorvastatin in Patients with Hypertension and Dyslipidemia: In a double-blind, placebo-controlled study of 1,660 patients with comorbid hypertension and dyslipidemia, once-daily treatment with eight dose combinations of amlodipine and atorvastatin (5/10 mg, 10/10 mg, 5/20 mg, 10/20 mg, 5/40 mg, 10/40 mg, 5/80 mg, or 10/80 mg) was compared vs. amlodipine alone (5 mg or 10 mg), atorvastatin alone (10 mg, 20 mg, 40 mg, or 80 mg), and placebo. In addition to concomitant hypertension and dyslipidemia, 15% of the patients had diabetes mellitus, 22% were smokers and 14% had a positive family history of CVD. At 8 weeks, all eight combination-treatment groups demonstrated statistically significant dose-related reductions in systolic blood pressure (SBP), diastolic blood pressure (DBP) and LDL-C compared to placebo, with no overall modification of effect of either component on SBP, DBP and LDL-C (see table as follows).
Efficacy in Terms of Reduction in Blood Pressure and LDL-C: See Tables 1, 2 and 3.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In an open-label trial, 1,220 patients with comorbid hypertension and dyslipidemia received elective dose titration with Amlodipine besilate/Atorvastatin calcium over a 14-week period. Patients were required to have uncontrolled BP to enter the trial (whether or not they were using antihypertensive medications at enrollment; patients were allowed to continue on previous antihypertensives, other than calcium channel blockers, during the 14-week dose titration period) but could enter with either controlled or uncontrolled LDL-C. As a result, no patient entered the trial with both BP and LDL-C controlled, and neither was controlled in 62% of patients. Treatment with Amlodipine besilate/Atorvastatin calcium reduced mean BP -17.1 mmHg systolic and -9.6 mmHg diastolic, and reduced mean LDL-C by -32.7%, resulting in control of both BP and LDL-C for 58% of these patients (controlled BP and LDL-C were defined, respectively, as <140/90 mmHg and <160 mg/dL for patients with comorbid hypertension and dyslipidemia only; <140/90 mmHg and <130 mg/dL for patients with comorbid hypertension and dyslipidemia plus 1 additional cardiovascular risk factor, excluding known CHD or diabetes mellitus; and <130/85 mmHg and <100 mg/dL for patients with comorbid hypertension and dyslipidemia plus known CHD, diabetes mellitus, or other atherosclerotic disease). Only 13% of the patients in this trial used Amlodipine besilate/Atorvastatin calcium as initial therapy for comorbid hypertension and dyslipidemia, whereas the amlodipine component of Amlodipine besilate/Atorvastatin calcium comprised add-on therapy for hypertension in 56% of patients, including patients for whom the atorvastatin calcium component of Amlodipine besilate/Atorvastatin calcium comprised initial therapy for dyslipidemia (20%), a substitution for atorvastatin taken previously (18%), or a switch from another statin (18%). When evaluated according to the use of antihypertensive and lipid-lowering medications at enrollment, results showed that both BP and LDL-C were brought under control for 65% of patients who used Amlodipine besilate/Atorvastatin calcium as initial therapy for comorbid hypertension and dyslipidemia and for 55% to 64% of patients for whom the amlodipine component of Amlodipine besilate/Atorvastatin calcium constituted add-on therapy for hypertension (55% for such patients who had previously used lipid-lowering medications other than atorvastatin, 58% for such patients who had previously used atorvastatin, and 64% for such patients who had not previously used lipid lowering medications).
Anglo-Scandinavian Cardiac Outcomes Trial: The Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) is a randomized 2x2 factorial design study comparing two antihypertensive regimens in a total of 19,342 patients (Blood Pressure Lowering arm - ASCOT-BPLA), as well as the effect of addition of 10 mg atorvastatin compared to placebo in 10,305 patients (Lipid-Lowering arm - ASCOT-LLA) on fatal and non-fatal coronary events. There are 19,257 and 10,240 efficacy evaluable patients in ASCOT-BPLA and ASCOT-LLA, respectively.
In Anglo-Scandinavian Cardiac Outcomes Trial Blood Pressure Lowering Arm: The effect of treatment regimens based on amlodipine (5 mg-10 mg) (n = 9681) or atenolol (50 mg-100 mg) (n = 9661) was compared in a prospective randomized open blinded endpoint (PROBE) design in 19,342 hypertensive patients, ≥40 to <80 years of age with no previous MI or treatment for angina, at least three of the following predefined cardiovascular risk factors: male gender, age ≥55 years, smoking, Type 2 diabetes, history of CAD event occurring in a first-degree relative before the age of 55 years (males) or 60 years (females), total-C: HDL ≥6, peripheral vascular disease, left ventricular hypertrophy, prior cerebrovascular event, specific electrocardiogram (ECG) abnormalities, proteinuria/albuminuria.
To attain further BP goals (<140/90 mm Hg for non-diabetic patients, <130/80 mm Hg for diabetic patients), perindopril (4 mg-8 mg) could be added to the amlodipine group and bendroflumethiazide potassium (1.25 mg-2.5 mg) to the atenolol group. Third line therapy was doxazosin gastrointestinal therapeutic system (GITS) (4 mg-8 mg) in both arms.
The ASCOT-BPLA study was stopped prematurely after 903 primary events (non-fatal MI and fatal CHD) with median follow-up of 5.5 years due to significant benefit of the amlodipine based regimen on the following secondary endpoints: all-cause mortality, cardiovascular (CV) mortality and stroke. The study had planned to need at least 1,150 primary endpoints.
The primary endpoint of non-fatal MI + fatal CHD did not reach statistical significance when comparing the amlodipine-based group to the atenolol-based group. The secondary endpoints of total coronary events, all-cause mortality, fatal and non-fatal stroke were statistically significantly reduced when comparing amlodipine-based group to the atenolol-based group.
The incidence of the primary and secondary endpoints in the 19,257 efficacy evaluable patients: See Table 4.
Click on icon to see table/diagram/image
Blood pressure (SBP/DBP) decreased significantly on both treatment regimens when compared to baseline (p-values <0.001). The SBP/DBP decreases from baseline were significantly more with the amlodipine based regimen than with the atenolol-based regimen (-27.5/-17.7 mmHg vs. -25.7/-15.6 mmHg, respectively), and the p-values on differences between the two groups were both <0.001 for SBP and DBP.
In Anglo-Scandinavian Cardiac Outcomes Trial Lipid-Lowering Arm: In the ASCOT-LLA, the effect of atorvastatin on fatal and non-fatal CHD was assessed in 10,305 hypertensive patients 40 to 80 years of age (mean of 63 years), without a previous MI and with TC levels <6.5 mmol/L (251 mg/dL). Additionally all patients had at least three of the following cardiovascular risk factors: male gender, age >55 years, smoking, diabetes, history of CHD in a first-degree relative, TC:HDL >6, peripheral vascular disease, left ventricular hypertrophy, prior cerebrovascular event, specific ECG abnormality, proteinuria/albuminuria. In this double-blind, placebo-controlled study patients were treated with antihypertensive therapy (goal BP <140/90 mmHg for non-diabetic patients, <130/80 mmHg for diabetic patients) and allocated to either atorvastatin 10 mg daily (n = 5168) or placebo (n = 5137). As the effect of atorvastatin treatment compared to placebo exceeded the significance threshold during an interim analysis, the ASCOT-LLA was terminated early at 3.3 years instead of 5 years. Additionally, BP was well controlled and similar in patients assigned to atorvastatin and placebo. These changes persisted throughout the treatment period.
Atorvastatin reduced the rate of the following events: See Table 5.
Click on icon to see table/diagram/image
The total mortality and cardiovascular mortality have not been significantly reduced, although a favorable trend was observed.
In Anglo-Scandinavian Cardiac Outcomes Trial 2x2: The pre-specified ASCOT 2x2 factorial analysis investigated the potential differential effect (interaction) of adding atorvastatin to the amlodipine vs. the atenolol group in ASCOT-LLA.
For the 10,305 patients enrolled in ASCOT-LLA, there were 5,168 patients in the atorvastatin group (2,584 patients received amlodipine and 2,584 patients received atenolol) and 5,137 in the placebo group (2,554 patients received amlodipine and 2,583 patients received atenolol).
The risk reductions on the composite endpoint of non-fatal MI and fatal CHD were based on the 10,240 efficacy evaluable patients.
The combination of amlodipine with atorvastatin resulted in a significant risk reduction in the composite primary endpoint of fatal CHD and non-fatal MI by: 53% (95% CI 31%-68%, p <0.0001) compared to amlodipine + placebo, 39% (95% CI 8%-59%, p <0.016) compared to atenolol + atorvastatin.
The p-value for the interaction was 0.027 which was not statistically significant at the pre-specified 0.01 level.
Blood pressure (SBP/DBP) decreased significantly on all four treatment regimens when compared to baseline (p-values <0.001). The SBP/DBP decreases from baseline were significantly more with the amlodipine based regimens than with the atenolol based regimens (-26.5/-15.6 mmHg vs. - 24.7/- 13.6 mmHg for amlodipine/atorvastatin vs. atenolol/atorvastatin, and -27.1/-15.8 mmHg vs. - 24.1/- 13.6 mmHg for amlodipine/placebo vs. atenolol/placebo, respectively). The p-values on differences between the two groups were all <0.01 for SBP and DBP.
Amlodipine Pharmacodynamics: Amlodipine is a calcium ion influx inhibitor (slow channel blocker or calcium ion antagonist) and inhibits the transmembrane influx of calcium ions into cardiac and vascular smooth muscle.
The mechanism of the antihypertensive action of amlodipine is due to a direct relaxant effect on vascular smooth muscle. The precise mechanism by which amlodipine relieves angina has not been fully determined but amlodipine reduces total ischemic burden by the following two actions.
Amlodipine dilates peripheral arterioles and thus, reduces the total peripheral resistance (afterload) against which the heart works. Since the heart rate remains stable, this unloading of the heart reduces myocardial energy consumption and oxygen requirements.
The mechanism of action of amlodipine also probably involves dilatation of the main coronary arteries and coronary arterioles, both in normal and ischemic regions. This dilatation increases myocardial oxygen delivery in patients with coronary artery spasm (Prinzmetal's or variant angina) and blunts smoking-induced coronary vasoconstriction.
In patients with hypertension, once daily dosing provides clinically significant reductions of BP in both the supine and standing positions throughout the 24 hour interval. Due to the slow onset of action, acute hypotension is not a feature of amlodipine administration.
In patients with angina, once daily administration of amlodipine increases total exercise time, time to angina onset, and time to 1 mm ST segment depression, and decreases both angina attack frequency and nitroglycerine tablet consumption.
Amlodipine has not been associated with any adverse metabolic effects or changes in plasma lipids and is suitable for use in patients with asthma, diabetes, and gout.
Use in Patients with CAD: The effects of amlodipine on cardiovascular morbidity and mortality, the progression of coronary atherosclerosis, and carotid atherosclerosis were studied in the Prospective Randomized Evaluation of the Vascular Effects of NORVASC Trial (PREVENT). This multicenter, randomized, double blind, placebo-controlled study followed 825 patients with angiographically defined CAD for 3 years. The population included patients with previous MI (45%), percutaneous transluminal coronary angioplasty (PTCA) at baseline (42%), or history of angina (69%). Severity of CAD ranged from 1-vessel disease (45% of patients) to 3+ vessel disease (21% of patients). Patients with uncontrolled hypertension (DBP > 95 mm Hg) were excluded from the study. Major cardiovascular events (MCVE) were adjudicated by a blinded endpoint committee. Although there were no demonstrable effects on the rate of progression of coronary artery lesions, amlodipine arrested the progression of carotid intima-media thickening. A significant reduction (- 31%) was observed in the amlodipine-treated patients in the combined endpoint of cardiovascular death, MI, stroke, PTCA, coronary artery bypass graft (CABG), hospitalization for unstable angina, and worsening CHF. A significant reduction (-42%) in revascularization procedures (PTCA and CABG) was also seen in the amlodipine-treated patients. Fewer hospitalizations (-33%) were seen for unstable angina in amlodipine-treated patients than in the placebo group.
The effectiveness of amlodipine in preventing clinical events in patients with CAD has been evaluated in an independent, multicenter, randomized, double-blind, placebo-controlled study of 1,997 patients; Comparison of Amlodipine vs. Enalapril to Limit Occurrences of Thrombosis (CAMELOT). Of these, 663 were treated with amlodipine 5 mg to 10 mg and 655 patients were treated with placebo, in addition to standard care of statins, beta-blockers, diuretics and aspirin, for 2 years. The key efficacy results are presented in Table 6. The results indicate that amlodipine treatment was associated with fewer hospitalizations for angina and revascularization procedures in patients with CAD. (See Table 6.)
Click on icon to see table/diagram/image
Treatment to Prevent Heart Attack Trial: A randomized double-blind morbidity-mortality study called the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) was performed to compare newer drug therapies: amlodipine 2.5 mg to 10 mg/day (calcium channel blocker) or lisinopril 10 mg to 40 mg/day (ACE-inhibitor) as first-line therapies to that of the thiazide-diuretic, chlorthalidone 12.5 to 25 mg/day in mild to moderate hypertension.
A total of 33,357 hypertensive patients aged 55 or older were randomized and followed for a mean of 4.9 years. The patients had at least one additional CHD risk factor, including MI or stroke >6 months or documentation of other atherosclerotic CVD (overall 51.5%), type 2 diabetes (36.1%), HDL-C <35 mg/dL (11.6%), left ventricular hypertrophy diagnosed by electrocardiogram or echocardiography (20.9%), current cigarette smoking (21.9%).
The primary endpoint was a composite of fatal CHD or non-fatal MI. There was no significant difference in the primary endpoint between amlodipine-based therapy and chlorthalidone-based therapy: RR 0.98; 95% CI 0.90-1.07; p = 0.65. In addition, there was no significant difference in all-cause mortality between amlodipine-based therapy and chlorthalidone-based therapy: RR 0.96; 95% CI 0.89-1.02; p = 0.20.
Use in Patients with Heart Failure: Hemodynamic studies and exercise based controlled clinical trials in NYHA Class II-IV heart failure patients have shown that amlodipine did not lead to clinical deterioration as measured by exercise tolerance, left ventricular ejection fraction and clinical symptomatology.
A placebo-controlled study (PRAISE) designed to evaluate patients in NYHA Class III-IV heart failure receiving digoxin, diuretics and ACE inhibitors has shown that amlodipine did not lead to an increase in risk of mortality or combined mortality and morbidity in patients with heart failure.
In a follow-up, long-term, placebo controlled study (PRAISE-2) of amlodipine in patients with NYHA III - IV heart failure without clinical symptoms or objective findings suggestive of underlying ischemic disease, on stable doses of ACE inhibitors, digitalis, and diuretics, amlodipine had no effect on total or cardiovascular mortality. In this same population amlodipine was associated with increased reports of pulmonary edema despite no significant difference in the incidence of worsening heart failure as compared to placebo.
Use in Pediatric Patients (Aged 6 to 17 years): The efficacy of amlodipine in hypertensive pediatric patients 6 to 17 years of age was demonstrated in one 8-week double-blind, placebo-controlled randomized withdrawal trial in 268 patients with hypertension. All patients were randomized to the 2.5 mg or 5 mg treatment arms and followed for 4 weeks after which they were randomized to continue 2.5 mg or 5 mg amlodipine or placebo for an additional 4 weeks. Compared to baseline, once daily treatment with amlodipine 5 mg resulted in statistically significant reductions in SBP and DBP. Placebo-adjusted, mean reduction in seated SBP was estimated to be 5.0 mmHg for the 5 mg dose of amlodipine and 3.3 mmHg for the 2.5 mg dose of amlodipine. Subgroup analyses indicated that younger pediatric patients aged 6 to 13 years had efficacy results comparable to those of the older pediatric patients aged 14 to 17 years.
Atorvastatin Pharmacodynamics: Atorvastatin is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts HMG-CoA to mevalonate, a precursor of sterols, including cholesterol. In patients with homozygous and heterozygous FH, nonfamilial forms of hypercholesterolemia, and mixed dyslipidemia, atorvastatin reduces total-C, LDL-C, and apo B. Atorvastatin also reduces very-low-density lipoprotein cholesterol (VLDL-C) and TG and produces variable increases in HDL-C.
Atorvastatin lowers plasma cholesterol and lipoprotein levels by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and by increasing the number of hepatic LDL receptors on the cell surface for enhanced uptake and catabolism of LDL.
Atorvastatin reduces LDL production and the number of LDL particles. Atorvastatin produces a profound and sustained increase in LDL receptor activity coupled with a beneficial change in the quality of circulating LDL particles. Atorvastatin is effective in reducing LDL in patients with homozygous FH, a population that has not normally responded to lipid-lowering medication.
Atorvastatin and some of its metabolites are pharmacologically active in humans. The primary site of action of atorvastatin is the liver, which is the principal site of cholesterol synthesis and LDL clearance. LDL-C reduction correlates better with drug dose than it does with systemic drug concentration. Individualization of drug dosage should be based on therapeutic response (see Dosage & Administration).
In a dose-response study, atorvastatin (10-80 mg) reduced total-C (30%-46%), LDL-C (41%-61%), apo B (34%-50%), and TG (14%-33%). These results are consistent in patients with heterozygous FH, nonfamilial forms of hypercholesterolemia, and mixed hyperlipidemia, including patients with non-insulin-dependent diabetes mellitus.
In patients with isolated hypertriglyceridemia, atorvastatin reduces total-C, LDL-C, VLDL-C, apo B, TG, and non-HDL-C, and increases HDL-C. In patients with dysbetalipoproteinemia, atorvastatin reduces intermediate density lipoprotein cholesterol (IDL-C).
In patients with Fredrickson Types IIa and IIb hyperlipoproteinemia pooled from 24 controlled trials, the median percent increases from baseline in HDL-C for atorvastatin (10-80 mg) were 5.1% to 8.7% in a non-dose-related manner. Additionally, analysis of this pooled data demonstrated significant dose related decreases in total-C/HDL-C and LDL-C/HDL-C ratios, ranging from -29% to -44% and -37% to -55%, respectively.
The effects of atorvastatin on ischemic events and total mortality were studied in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering study (MIRACL). This multicenter, randomized, double-blind, placebo-controlled study followed 3,086 patients with acute coronary syndromes; unstable angina or non-Q wave MI. Patients were treated with standard care, including diet, and either atorvastatin 80 mg daily or placebo for a median duration of 16 weeks. The final LDL-C, total-C, HDL-C and TG levels were 72 mg/dL, 147 mg/dL, 48 mg/dL, and 139 mg/dL in the atorvastatin group, respectively, and 135 mg/dL, 217 mg/dL, 46 mg/dL, and 187 mg/dL, respectively, in the placebo group. Atorvastatin significantly reduced the risk of ischemic events and death by 16%. The risk of experiencing re-hospitalization for angina pectoris with documented evidence of myocardial ischemia was significantly reduced by 26%. Atorvastatin reduced the risk of ischemic events and death to a similar extent across the range of baseline LDL-C. In addition, atorvastatin reduced the risk of ischemic events and death to similar extents in patients with non-Q wave MI and unstable angina, as well as in males and females and in patients ≤65 years of age and >65 years of age.
Prevention of Cardiovascular Complications: The effect of atorvastatin on fatal and non-fatal CHD is discussed in this section under Clinical Studies of Combined Amlodipine and Atorvastatin in Patients with Hypertension and Dyslipidemia, Anglo-Scandinavian Cardiac Outcomes Trial.
In the Collaborative Atorvastatin Diabetes Study (CARDS), the effect of atorvastatin on fatal and non-fatal CVD was assessed in 2838 patients with Type 2 diabetes 40 to 75 years of age, without prior history of CVD and with LDL ≤4.14 mmol/L (160 mg/dL) and TG ≤6.78 mmol/L (600 mg/dL). Additionally, all patients had at least one of the following risk factors: hypertension, current smoking, retinopathy, microalbuminuria or macroalbuminuria.
In this randomized, double-blind, multicenter, placebo-controlled trial, patients were treated with either atorvastatin 10 mg daily (n = 1428) or placebo (n = 1410) for a median follow-up of 3.9 years. As the effect of atorvastatin treatment on the primary endpoint reached the predefined stopping rules for efficacy, CARDS was terminated 2 years earlier than anticipated.
The absolute and relative risk reduction effect of atorvastatin is as follows: See Table 7.
Click on icon to see table/diagram/image
There was no evidence of a difference in the treatment effect by patient's gender, age, or baseline LDL-C level.
A relative risk reduction in death of 27% (82 deaths in the placebo group compared to 61 deaths in the treatment arm) has been observed with a borderline statistical significance (p = 0.0592).
The overall incidence of adverse events or serious adverse events was similar between the treatment groups.
Atherosclerosis: In the Reversing Atherosclerosis with Aggressive Lipid-Lowering Study (REVERSAL), the effect of atorvastatin 80 mg and pravastatin 40 mg on coronary atherosclerosis was assessed by intravascular ultrasound (IVUS), during angiography, in patients with CHD. In this randomized, double-blind, multicenter, controlled clinical trial, IVUS was performed at baseline and at 18 months in 502 patients. In the atorvastatin group (n = 253), the median percent change, from baseline, in total atheroma volume (the primary study criteria) was -0.4% (p = 0.98) in the atorvastatin group and +2.7% (p = 0.001) in the pravastatin group (n = 249). When compared to pravastatin, the effects of atorvastatin were statistically significant (p = 0.02).
In the atorvastatin group, LDL-C was reduced to a mean of 2.04 mmol/L ± 0.8 (78.9 mg/dL ± 30) from baseline 3.89 mmol/L ± 0.7 (150 mg/dL ± 28) and in the pravastatin group, LDL-C was reduced to a mean of 2.85 mmol/L ± 0.7 (110 mg/dL ± 26) from baseline 3.89 mmol/L ± 0.7 (150 mg/dL ± 26) (p<0.0001). Atorvastatin also significantly reduced mean TC by 34.1% (pravastatin: -18.4%, p<0.0001), mean TG levels by 20% (pravastatin: -6.8%, p<0.0009), and mean apo B by 39.1% (pravastatin: -22.0%, p<0.0001). Atorvastatin increased mean HDL-C by 2.9% (pravastatin: +5.6%, p = NS). There was a 36.4% mean reduction in CRP in the atorvastatin group compared to a 5.2% reduction in the pravastatin group (p<0.0001).
The safety and tolerability profiles of the two treatment groups were comparable.
Recurrent Stroke: In the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study, the effect of atorvastatin 80 mg daily or placebo on stroke was evaluated in 4,731 patients who had a stroke or TIA within the preceding 6 months and no history of CHD. Patients were 60% male, 21 to 92 years of age (average age 63 years), and had an average baseline LDL of 133 mg/dL (3.4 mmol/L). The mean LDL-C was 73 mg/dL (1.9 mmol/L) during treatment with atorvastatin and 129 mg/dL (3.3 mmol/L) during treatment with placebo. Median follow-up was 4.9 years.
Atorvastatin 80 mg reduced the risk of the primary endpoint of fatal or non-fatal stroke by 15% (hazard ratio [HR] 0.85; 95% CI 0.72-1.00; p = 0.05 or HR 0.84; 95% CI 0.71-0.99; p = 0.03 after adjustment for baseline factors) compared to placebo. Atorvastatin 80 mg significantly reduced the risk of major coronary events (HR 0.67; 95% CI 0.51-0.89; p = 0.006), any CHD event (HR 0.60; 95% CI 0.48-0.74; p<0.001), and revascularization procedures (HR 0.57; 95% CI 0.44-0.74; p<0.001).
In a post-hoc analysis, atorvastatin 80 mg, reduced the incidence of ischemic stroke (218/2365, 9.2% vs. 274/2366, 11.6%, p = 0.01) and increased the incidence of hemorrhagic stroke (55/2365, 2.3% vs. 33/2366, 1.4%, p = 0.02) compared to placebo. The incidence of fatal hemorrhagic stroke was similar between the groups (17 atorvastatin vs. 18 placebo). Reduction in the risk of cardiovascular events with atorvastatin 80 mg was demonstrated in all patient groups except in patients who entered the study with a hemorrhagic stroke and had a recurrent hemorrhagic stroke (7 atorvastatin vs. 2 placebo), where the number of events was too small to discern risk or benefit.
In patients treated with atorvastatin 80 mg, there were fewer strokes of any type (265 atorvastatin vs. 311 placebo) and fewer CHD events (123 atorvastatin vs. 204 placebo). Overall mortality was similar across treatment groups (216 atorvastatin vs. 211 placebo). The overall incidence of adverse events and serious adverse events was similar between the treatment groups.
Secondary Prevention of Cardiovascular Events: In the Treating to New Targets Study (TNT), the effect of atorvastatin 80 mg/day vs. atorvastatin 10 mg/day on the reduction in cardiovascular events was assessed in 10,001 subjects (94% white, 81% male, 38% ≥65 years) with clinically evident CHD who had achieved a target LDL-C level <130 mg/dL after completing an 8-week, open-label, run-in period with atorvastatin 10 mg/day. Subjects were randomly assigned to either 10 mg/day or 80 mg/day of atorvastatin and followed for a median duration of 4.9 years. The mean LDL-C, TC, TG, non-HDL and HDL cholesterol levels at 12 weeks were 73 mg/dL, 145 mg/dL, 128 mg/dL, 98 mg/dL and 47 mg/dL, respectively, during treatment with 80 mg of atorvastatin and 99 mg/dL, 177 mg/dL, 152 mg/dL, 129 mg/dL and 48 mg/dL, respectively, during treatment with 10 mg of atorvastatin.
Treatment with atorvastatin 80 mg/day significantly reduced the rate of MCVE (434 events in the 80 mg/day group vs. 548 events in the 10 mg/day group) with a relative risk reduction of 22%.
Atorvastatin 80 mg significantly reduced the risk of the following: See Table 8.
Click on icon to see table/diagram/image
There was no significant difference between the treatment groups for all-cause mortality: 282 (5.6%) in the atorvastatin 10 mg/day group vs. 284 (5.7%) in the atorvastatin 80 mg/day group. The proportions of subjects who experienced cardiovascular death, including the components of CHD death and fatal stroke were numerically smaller in the atorvastatin 80 mg group than in the atorvastatin 10 mg treatment group. The proportions of subjects who experienced non-cardiovascular death were numerically larger in the atorvastatin 80 mg group than in the atorvastatin 10 mg treatment group.
In the Incremental Decrease in Endpoints Through Aggressive Lipid Lowering Study (IDEAL), treatment with atorvastatin 80 mg/day was compared to treatment with simvastatin 20 mg/day to 40 mg/day in 8,888 subjects up to 80 years of age with a history of CHD to assess whether reduction in CV risk could be achieved. Patients were mainly male (81%), white (99%) with an average age of 61.7 years and an average LDL-C of 121.5 mg/dL at randomization; 76% were on statin therapy. In this prospective, randomized, open-label, blinded endpoint (PROBE) trial with no run-in period, subjects were followed for a median duration of 4.8 years. The mean LDL-C, TC, TG, HDL and non-HDL cholesterol levels at Week 12 were 78 mg/dL, 145 mg/dL, 115 mg/dL, 45 mg/dL and 100 mg/dL, respectively, during treatment with 80 mg of atorvastatin and 105 mg/dL, 179 mg/dL, 142 mg/dL, 47 mg/dL and 132 mg/dL, respectively, during treatment with 20 mg to 40 mg of simvastatin.
There was no significant difference between the treatment groups for the primary endpoint, the rate of first major coronary event (fatal CHD, non-fatal MI and resuscitated cardiac arrest): 411 (9.3%) in the atorvastatin 80 mg/day group vs. 463 (10.4%) in the simvastatin 20 mg to 40 mg/day group, HR 0.89; 95% CI 0.78, 1.01; p = 0.07.
There were no significant differences between the treatment groups for all-cause mortality: 366 (8.2%) in the atorvastatin 80 mg/day group vs. 374 (8.4%) in the simvastatin 20 mg to 40 mg/day group. The proportions of subjects who experienced CV or non-CV death were similar for the atorvastatin 80 mg group and the simvastatin 20 mg to 40 mg group.
Heterozygous Familial Hypercholesterolemia in Pediatric Patients: The following pediatric-exclusive studies have been completed with atorvastatin.
In an open-label, single-arm study, 271 male and female Heterozygous Familial Hypercholesterolemia (HeFH) children 6-15 years of age were enrolled and treated with atorvastatin for up to 3 years. Inclusion in the study required confirmed HeFH and a baseline LDL-C level ≥4 mmol/L (approximately 152 mg/dL). The study included 139 children at Tanner 1 development stage (generally ranging from 6-10 years of age). The dosage of atorvastatin (once daily) was initiated at 5 mg (chewable tablet) in children less than 10 years of age. Children age 10 and above were initiated at 10 mg atorvastatin (once daily). All children could titrate to higher doses to achieve a target of <3.35 mmol/L LDL-C. The mean weighted dose for children aged 6 to 9 years was 19.6 mg and the mean weighted dose for children aged 10 years and above was 23.9 mg.
The mean (± SD) baseline LDL-C value was 6.12 (1.26) mmol/L which was approximately 233 (48) mg/dL. See Table 9 as follows for final results.
The data were consistent with no drug effect on any of the parameters of growth and development (i.e., height, weight, BMI, Tanner stage, Investigator assessment of Overall Maturation and Development) in pediatric and adolescent subjects with HeFH receiving atorvastatin treatment over the 3 year study. There was no Investigator-assessed drug effect noted in height, weight, BMI by age or by gender by visit. (See Table 9.)
Click on icon to see table/diagram/image
In a double-blind, placebo-controlled study followed by an open-label phase, 187 boys and postmenarchal girls 10 to 17 years of age (mean age 14.1 years) with heterozygous FH or severe hypercholesterolemia were randomized to atorvastatin (n = 140) or placebo (n = 47) for 26 weeks and then all received atorvastatin for 26 weeks. Inclusion in the study required a baseline LDL-C level ≥190 mg/dL or a baseline LDL-C ≥160 mg/dL and positive family history of FH or documented premature CVD in a first- or second-degree relative. The mean baseline LDL-C value was 218.6 mg/dL (range: 138.5-385.0 mg/dL) in the atorvastatin group compared to 230.0 mg/dL (range: 160.0-324.5 mg/dL) in the placebo group. The dosage of atorvastatin (once daily) was 10 mg for the first 4 weeks and up-titrated to 20 mg if the LDL-C level was >130 mg/dL. The number of atorvastatin-treated patients who required up-titration to 20 mg after Week 4 during the double-blind phase was 78 (55.7%).
Atorvastatin significantly decreased plasma levels of total-C, LDL-C, TG, and apo B during the 26 week double-blind phase (see Table 10).
Click on icon to see table/diagram/image
The mean achieved LDL-C value was 130.7 mg/dL (range: 70.0-242.0 mg/dL) in the atorvastatin group compared to 228.5 mg/dL (range: 152.0-385.0 mg/dL) in the placebo group during the 26 week double-blind phase. In this 1-year study, there was no detectable effect on growth or sexual maturation in boys or on menstrual cycle length in girls.
An 8-week, open-label study to evaluate pharmacokinetics, pharmacodynamics, and safety and tolerability of atorvastatin was conducted in 39 patients, 6 to 17 years of age with genetically confirmed heterozygous familial hypercholesterolemia and baseline LDL-C ≥4 mmol/L. Cohort A included 15 patients, 6 to 12 years of age and at Tanner Stage 1. Cohort B included 24 patients, 10 to 17 years of age and at Tanner Stage ≥2.
The initial dose of atorvastatin was 5 mg daily of a chewable tablet in Cohort A and 10 mg daily of a tablet formulation in Cohort B. The atorvastatin dose was permitted to be doubled if a patient had not attained target LDL-C of <3.35 mmol/L at Week 4 and if atorvastatin was well tolerated.
Mean values for LDL-C, TC, VLDL-C, and Apo B decreased by Week 2 among all patients. For patients whose dose was doubled, additional decreases were observed as early as 2 weeks, at the first assessment, after dose escalation. The mean percent decreases in lipid parameters were similar for both cohorts, regardless of whether patients remained at their initial dose or doubled their initial dose. At Week 8, on average, the percent change from baseline in LDL-C and TC was approximately 40% and 30%, respectively, over the range of exposures.
The long-term efficacy of atorvastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Pharmacokinetics: Absorption: In studies with amlodipine/atorvastatin: Following oral administration of Amlodipine besilate/Atorvastatin calcium (Norvasc Protect) two distinct peak plasma concentrations were observed. The first, within 1 to 2 hours of administration, is attributable to atorvastatin; the second, between 6 and 12 hours after dosing is attributable to amlodipine. The rate and extent of absorption (bioavailability) of amlodipine and atorvastatin from Amlodipine besilate/Atorvastatin calcium (Norvasc Protect) are not significantly different from the bioavailability of amlodipine and atorvastatin from co-administration of amlodipine and atorvastatin tablets as assessed by C
max: 101% (90% CI: 98, 104) and AUC: 100% (90% CI: 97, 103) for the amlodipine component and C
max: 94% (90% CI: 85, 104) and AUC: 105% (90% CI: 99, 111) for the atorvastatin component, respectively.
The bioavailability of the amlodipine component of Amlodipine besilate/Atorvastatin calcium (Norvasc Protect) was not affected under the fed state as assessed by C
max: 105% (90% CI: 99, 111) and AUC: 101% (90% CI: 97, 105) relative to the fasted state. Although food decreases the rate and extent of absorption of atorvastatin from Amlodipine besilate/Atorvastatin calcium (Norvasc Protect) by approximately 32% and 11%, respectively, as assessed by C
max: 68% (90% CI 60, 79) and AUC: 89% (90% CI 83, 95) relative to the fasted state, similar reductions in plasma concentrations in the fed state have been seen with atorvastatin taken as monotherapy without reduction in LDL-C effect (see as follows).
In studies with amlodipine: After oral administration of therapeutic doses, amlodipine is well absorbed with peak blood levels between 6 to 12 hours post-dose. Absolute bioavailability has been estimated to be between 64% and 80%. The volume of distribution is approximately 21 L/kg.
In vitro studies have shown that approximately 97.5% of circulating amlodipine is bound to plasma proteins.
Absorption of amlodipine is unaffected by consumption of food.
In studies with atorvastatin: Atorvastatin is rapidly absorbed after oral administration; maximum plasma concentrations occur within 1 to 2 hours. Extent of absorption and plasma atorvastatin concentrations increases in proportion to atorvastatin dose. Atorvastatin tablets are 95% to 99% bioavailable compared to solutions. The absolute bioavailability of atorvastatin is approximately 14% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism. Although food decreases the rate and extent of drug absorption by approximately 25% and 9% respectively, as assessed by C
max and AUC, LDL-C reduction is similar whether atorvastatin is given with or without food. Plasma atorvastatin concentrations are lower (approximately 30% for C
max and AUC) following evening drug administration compared to morning. However, LDL-C reduction is the same regardless of the time of day of drug administration (see Dosage & Administration).
Distribution: In studies with atorvastatin: Mean volume of distribution of atorvastatin is approximately 381 L. Atorvastatin is ≥98% bound to plasma proteins. A red blood cell/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells.
Metabolism and Excretion: In studies with amlodipine: The terminal plasma elimination half life is about 35 to 50 hours and is consistent with once daily dosing. Steady-state plasma levels are reached after 7 to 8 days of consecutive dosing. Amlodipine is extensively metabolized by the liver to inactive metabolites with 10% of the parent compound and 60% of metabolites excreted in the urine.
In studies with atorvastatin: Atorvastatin is extensively metabolized to ortho- and para-hydroxylated derivatives and various beta-oxidation products.
In vitro inhibition of HMG-CoA reductase by ortho- and para-hydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites.
In vitro studies suggest the importance of atorvastatin metabolism by hepatic cytochrome P450 3A4, consistent with increased plasma concentrations of atorvastatin in humans following co-administration with erythromycin, a known inhibitor of this isozyme.
In vitro studies also indicate that atorvastatin is a weak inhibitor of cytochrome P450 3A4. Atorvastatin co-administration did not produce a clinically significant effect in plasma concentrations of terfenadine, a compound predominantly metabolized by cytochrome P450 3A4; therefore, it is unlikely that atorvastatin will significantly alter the pharmacokinetics of other cytochrome P450 3A4 substrates (see Interactions). In animals, the ortho-hydroxy metabolite undergoes further glucuronidation.
Atorvastatin and its metabolites are eliminated primarily in bile following hepatic and/or extrahepatic metabolism; however, the drug does not appear to undergo enterohepatic recirculation. Mean plasma elimination half-life of atorvastatin in humans is approximately 14 hours, but the half-life of inhibitory activity for HMG-CoA reductase is 20 to 30 hours due to the contribution of active metabolites. Less than 2% of a dose of atorvastatin is recovered in urine following oral administration.
Atorvastatin is a substrate of the hepatic transporters, OATP1B1 and OATP1B3 transporter. Metabolites of atorvastatin are substrates of OATP1B1. Atorvastatin is also identified as a substrate of the efflux transporters MDR1 and BCRP, which may limit the intestinal absorption and biliary clearance of atorvastatin.
Special Populations: Hepatic Insufficiency: In studies with atorvastatin: Plasma concentrations of atorvastatin are markedly increased (approximately 16-fold in C
max and 11-fold in AUC) in patients with chronic alcoholic liver disease (Child-Pugh B) (see Contraindications).
Renal Insufficiency: See Dosage & Administration.
In studies with amlodipine: Changes in amlodipine plasma concentrations are not correlated with degree of renal impairment. Amlodipine is not dialyzable.
In studies with atorvastatin: Renal disease has no influence on the plasma concentrations or lipid effects of atorvastatin. Thus, dose adjustment in patients with renal dysfunction is not necessary.
Gender: In studies with atorvastatin: Plasma concentrations of atorvastatin in women differ (approximately 20% higher for C
max and 10% lower for AUC) from those in men. However, there were no clinically significant differences in lipid effects between men and women.
Elderly: In studies with amlodipine: The time to reach peak plasma concentrations of amlodipine is similar in elderly and younger subjects. Amlodipine clearance tends to be decreased with resulting increases in AUC and elimination half-life in elderly patients. Increases in AUC and elimination half life in patients with CHF were as expected for the patient age group studied. Amlodipine, used at similar doses in elderly or younger patients, is equally well tolerated.
In studies with atorvastatin: Plasma concentrations of atorvastatin are higher (approximately 40% for C
max and 30% for AUC) in healthy, elderly subjects (aged ≥65 years) than in young adults. The ACCESS study specifically evaluated elderly patients with respect to reaching their NCEP treatment goals. The study included 1,087 patients under 65 years of age, 815 patients over 65 years of age, and 185 patients over 75 years of age. No differences in safety, efficacy or lipid treatment goal attainment were observed between elderly patients and the overall population.
Pediatrics: In studies with amlodipine: In one clinical chronic exposure study, 73 hypertensive pediatric patients, aged 12 months to less than or equal to 17 years, amlodipine besilate was dosed at an average daily dose of 0.17 mg/kg. Clearance for subjects with the median weight of 45 kg was 23.7 L/h and 17.6 L/h for males and females, respectively. This is in a similar range to the published estimates of 24.8 L/h in a 70 kg adult. The average estimate for volume of distribution for a 45 kg patient was 1130 L (25.11 L/kg). Maintenance of the BP effect over the 24-hour dosing interval was observed with little difference in peak and trough variation effect. When compared to historical adult pharmacokinetics the parameters observed in this study indicate that once daily dosing is appropriate.
In studies with atorvastatin: In an open-label, 8-week study, Tanner Stage 1 (N = 15) and Tanner Stage ≥2 (N = 24) pediatric patients (ages 6-17 years) with heterozygous familial hypercholesterolemia and baseline LDL-C ≥4 mmol/L were treated with 5 or 10 mg of chewable or 10 or 20 mg of film-coated atorvastatin tablets once daily, respectively. Body weight was the only significant covariate in atorvastatin population PK model. Apparent oral clearance of atorvastatin in pediatric subjects appeared similar to adults when scaled allometrically by body weight. Consistent decreases in LDL-C and TC were observed over the range of atorvastatin and o-hydroxyatorvastatin exposures.
Drug Interactions: In studies with atorvastatin: The effect of co-administered drugs on the pharmacokinetics of atorvastatin as well as the effect of atorvastatin on the pharmacokinetics of co-administered drugs are summarized as follows (see Precautions and Interactions).
(See Tables 11 and 12.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Toxicology: Preclinical Safety Data: Carcinogenesis: In studies with amlodipine: Rats and mice treated with amlodipine in the diet for 2 years, at concentrations calculated to provide daily dosage levels of 0.5, 1.25, and 2.5 mg/kg/day showed no evidence of carcinogenicity. The highest dose (for mice, similar to, and for rats twice* the maximum recommended clinical dose of 10 mg on a mg/m
2 basis) was close to the maximum tolerated dose for mice but not for rats.
In studies with atorvastatin: Atorvastatin was not carcinogenic in rats. The maximum dose used was 63-fold higher than the highest human dose (80 mg/day) on a mg/kg body-weight basis and 8- to 16-fold higher based on AUC
(0-24) values. In a 2-year study in mice, incidences of hepatocellular adenomas in males and hepatocellular carcinomas in females were increased at the maximum dose used, which was 250-fold higher than the highest human dose on a mg/kg body-weight basis. Systemic exposure was 6- to 11-fold higher based on AUC
(0-24).
All other chemically similar drugs in this class have induced tumors in both mice and rats at multiples of 12 to 125 times their highest recommended clinical doses, on a mg/kg body weight basis.
*Based on patient weight of 50 kg.
Mutagenesis: In studies with amlodipine: Mutagenicity studies revealed no drug-related effects at either the gene or chromosome level.
In studies with atorvastatin: Atorvastatin did not demonstrate mutagenic or clastogenic potential in four
in vitro tests with and without metabolic activation or in one
in vivo assay. It was negative in the Ames test with
Salmonella typhimurium and
Escherichia coli, and in the
in vitro hypoxanthine-guanine phosphoribosyl transferase (HGPRT) forward mutation assay in Chinese hamster lung cells. Atorvastatin did not produce significant increases in chromosomal aberrations in the
in vitro Chinese hamster lung cell assay and was negative in the
in vivo mouse micronucleus test.
Impairment of Fertility: In studies with amlodipine: There was no effect on the fertility of rats treated with amlodipine (males for 64 days and females 14 days prior to mating) at doses up to 10 mg/kg/day (8 times* the maximum recommended human dose of 10 mg on a mg/m
2 basis).
*Based on patient weight of 50 kg.
In studies with atorvastatin: No adverse effects on fertility or reproduction were observed in male rats given doses of atorvastatin up to 175 mg/kg/day or in female rats given doses up to 225 mg/kg/day. These doses are 100 to 140 times the maximum recommended human dose on a mg/kg basis. Atorvastatin caused no adverse effects on sperm or semen parameters, or on reproductive organ histopathology in dogs given doses of 10 mg/kg, 40 mg/kg, or 120 mg/kg for 2 years.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image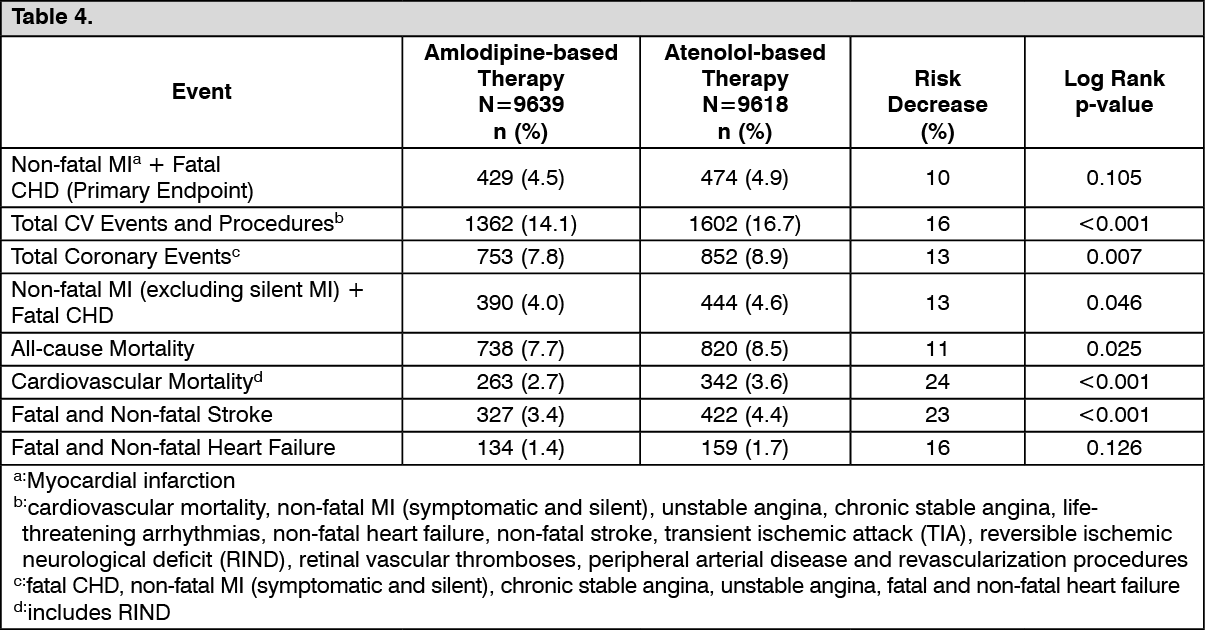 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image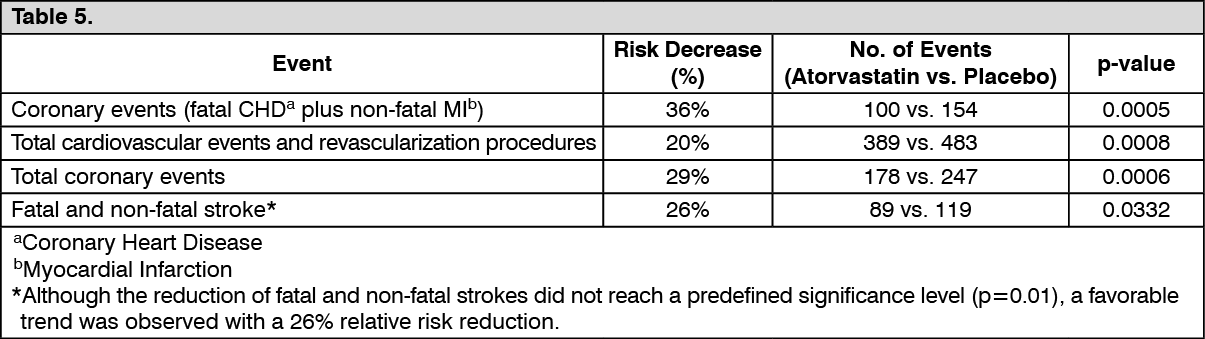 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image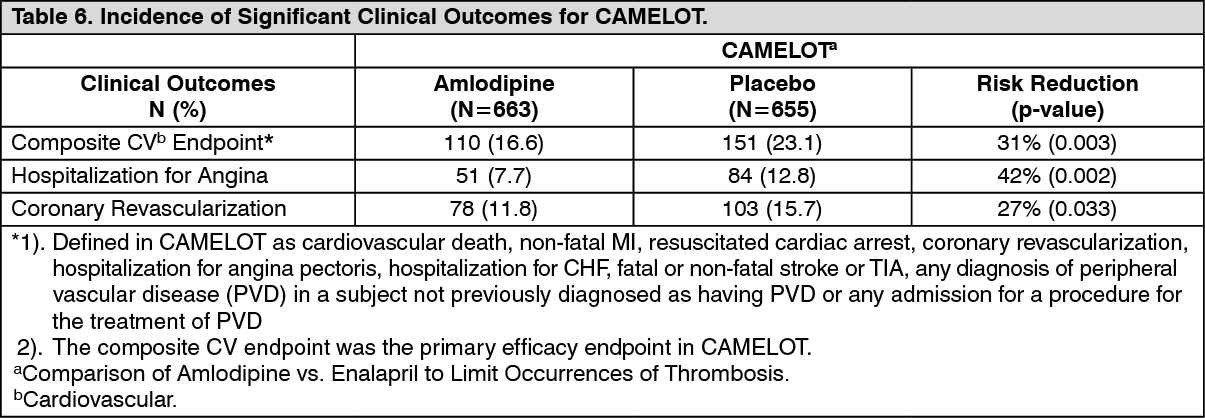 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image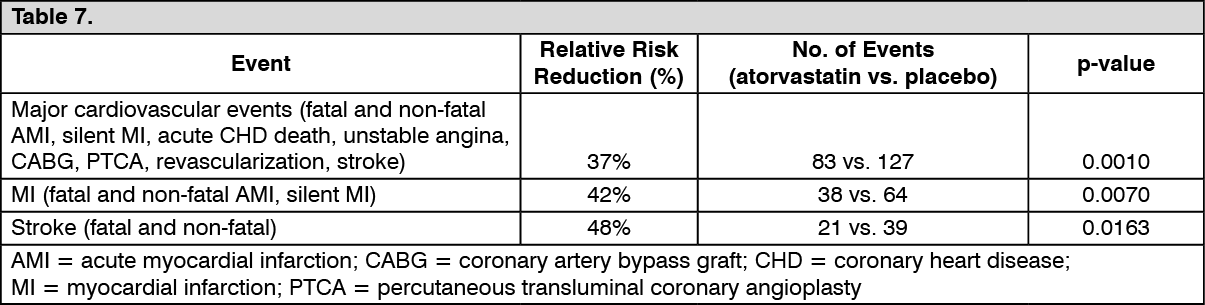 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image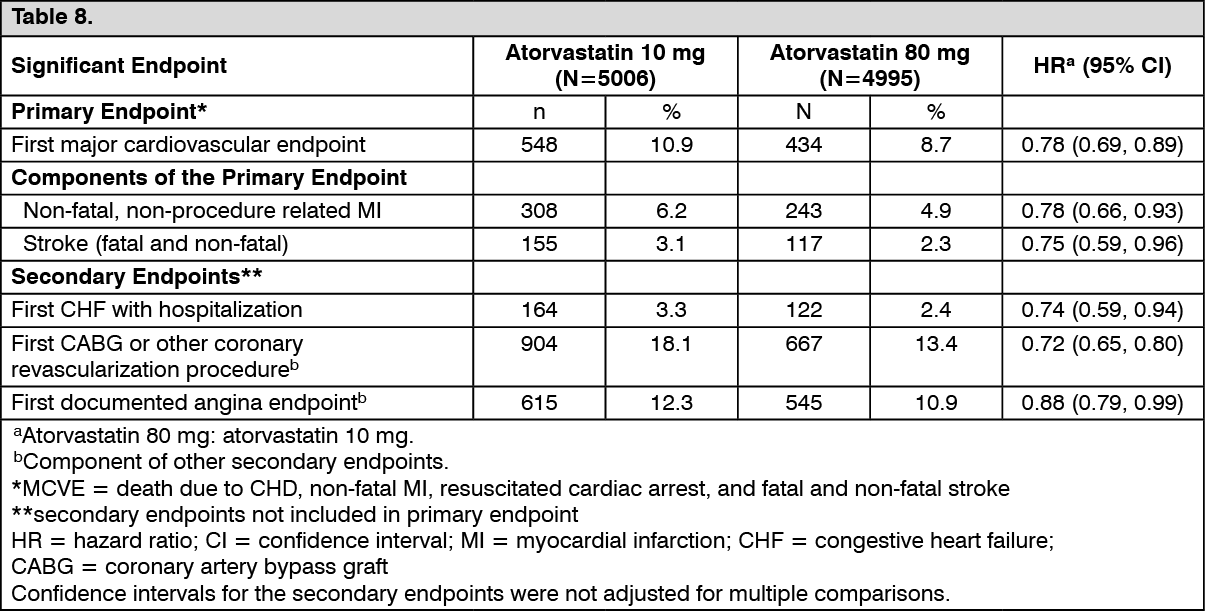 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image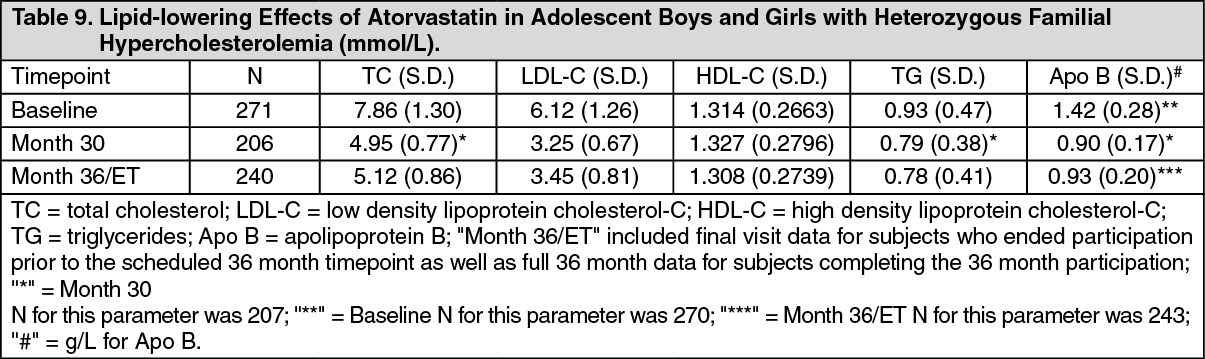 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image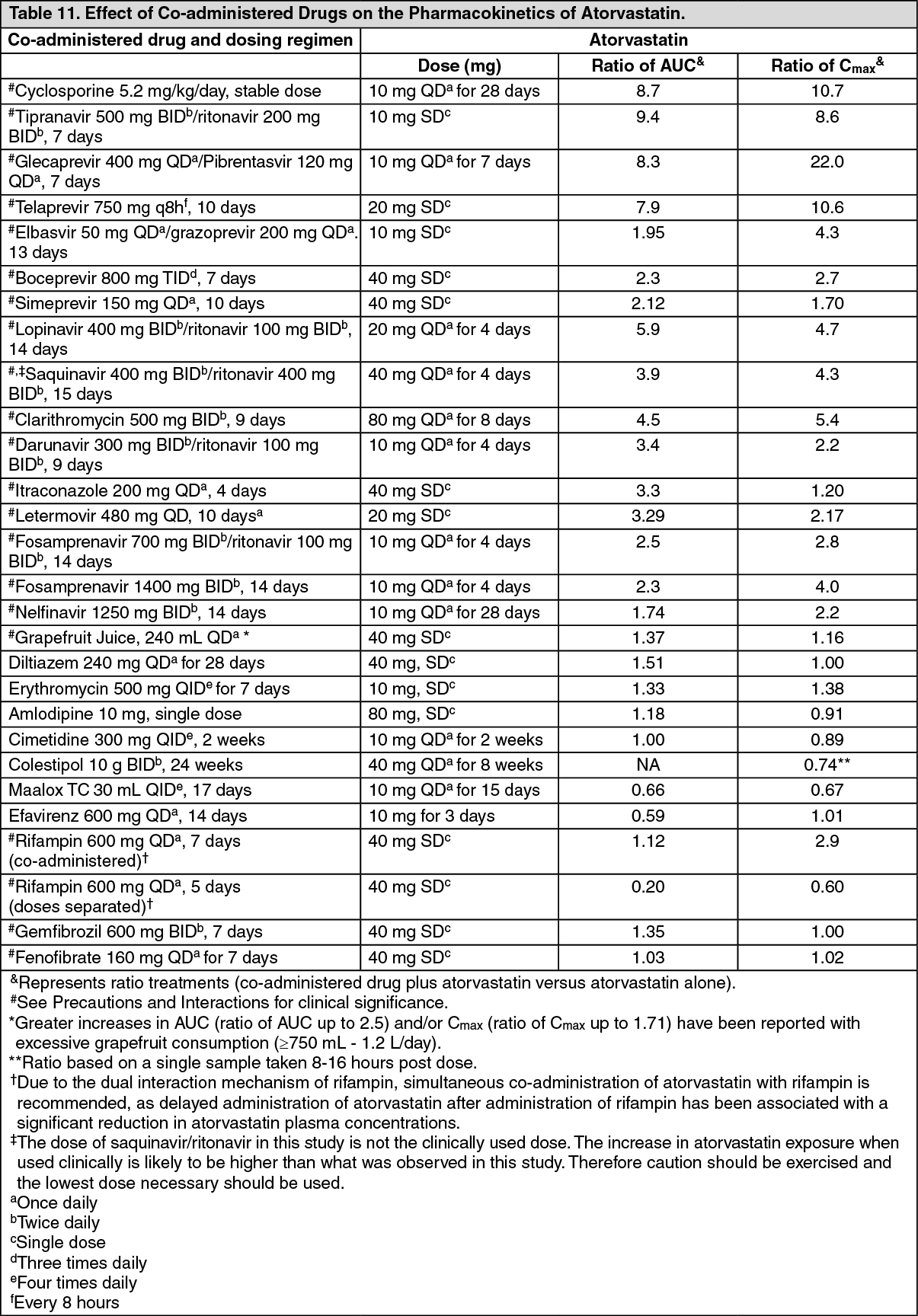 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image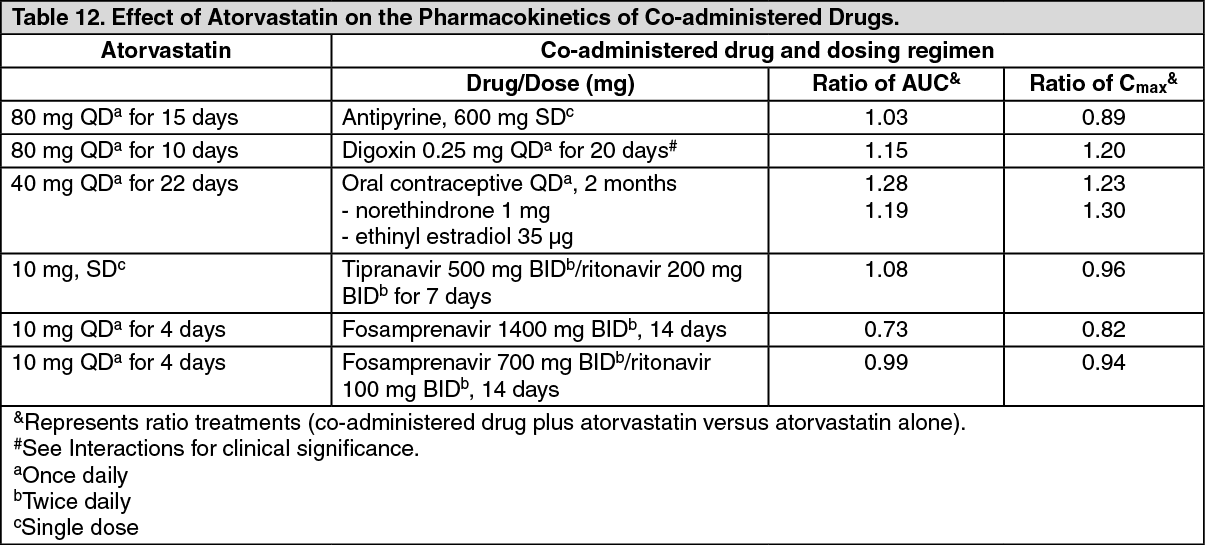 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image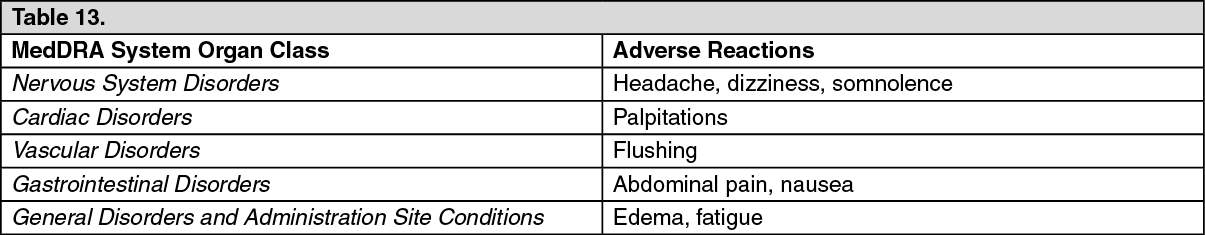 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image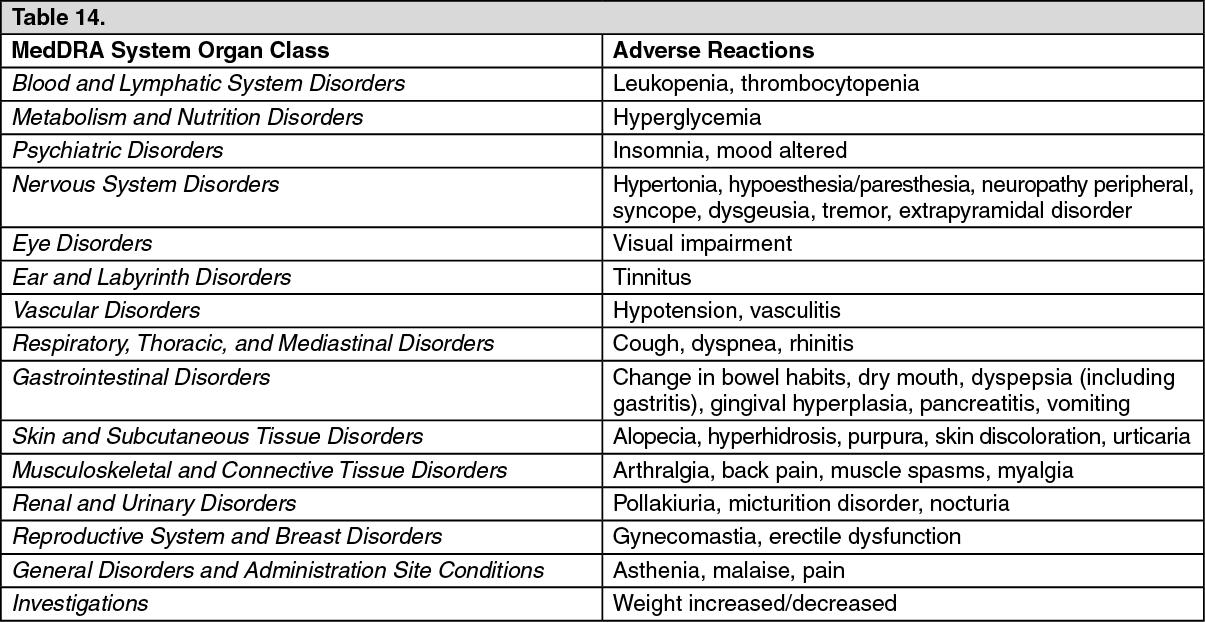 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image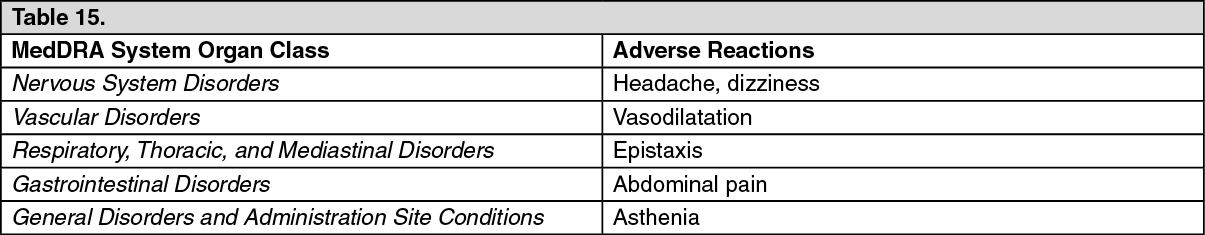 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out